Iniciativa de Vigilancia Genómica de SARS-CoV-2 de Yale
Esta semana añadimos 5 nuevos genomas de finales de Abril, los cuales son lo genomas más recientes de Connecticut (CT). Estos nuevos genomas, muestreados justo después del pico de contagios en CT (Abril 20-22), proveerán una percepción acerca de la transmisión que estuvo ocurriendo incluso con medidas estrictas de distanciamiento social.
Recapitulación
Previamente mostramos cómo los casos muestreados desde Marzo hasta principios de Abril estuvieron cercanamente relacionados a otros brotes domésticos en Estados Unidos.
Específicamente, 21 de nuestros primeros casos (en Marzo) estuvieron relacionados al SARS-CoV-2 que estuvo circulando en la Costa Oeste (encontrado en un grupo al cual lo denominamos clado-WA, debido al origen más temprano del grupo, ligado al estado de Washington). Este clado se encuentra dentro del linaje viral global ‘A’. Dentro de estos 21 genomas, existen 12 clusters de casos en CT que se encuentran más cercanamente conectados a los casos de WA que de otros en CT, lo que indica que existieron varias introducciones separadas de la costa oeste durante el inicio de la pandemia en CT. Esto es demostrado por nuestros datos de casos y viajes.
Linajes de SARS-CoV-2 en CT
| Linaje | Sub-linaje | Introducido a CT de | Fechas de muestreo | Número de secuencias |
| A | A.1 | Inicialmente introducido al Oeste de E.U.A../Canadá al menos dos veces, ahora existente en CT | Marzo 8 a Abril 9 | 21 |
| Ninguno | Asia/Oceanía al Noreste de E.U.A. | Marzo 13 a Abril 6 | 4 | |
| B | B.1 (más B.1.3 y B.1.1) | Nueva York | Marzo 11 a Abril 17 | 100 |
| B.2 | Sureste de Asia al noreste de E.U.A. | Marzo 6 a Abril 10 | 3 |
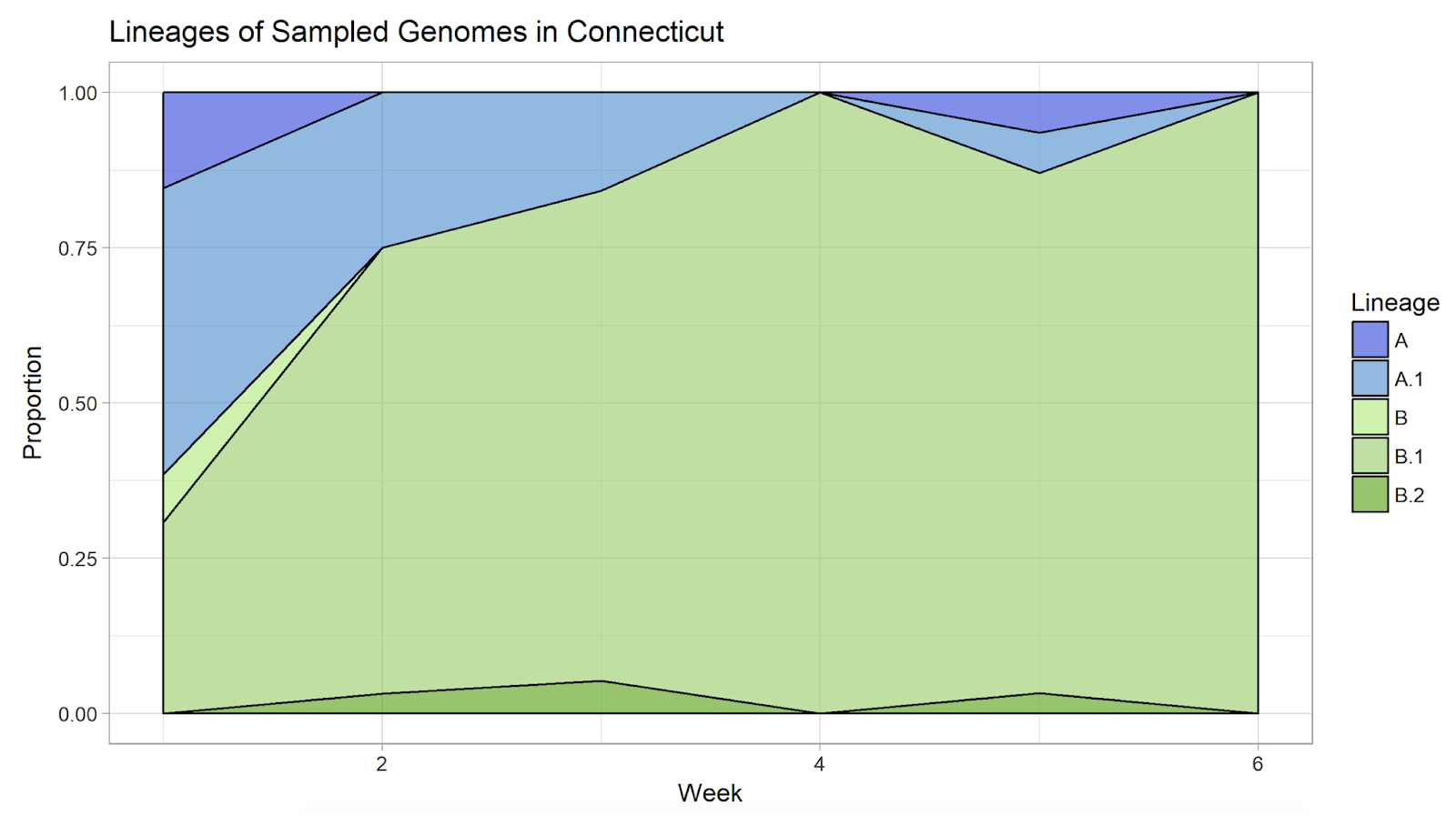
Adicionalmente, encontramos que durante Marzo hasta mediados de Abril, más y más virus de nuestras muestras se encontraban en el linaje ‘B.1’, el cual fue introducido primero a Nueva York (y por ello nombrado como clado-NY). Dentro de las 95 secuencias que obtuvimos dentro de este linaje, pareciera que existe una cantidad significativa de introducciones separadas seguidas por transmisión local a finales de Marzo/principios de Abril.
¿Qué hay de nuevo?
Añadimos 5 nuevas secuencias de finales de abril, las cuales son las primeras secuencias obtenidas después del pico de transmisión. Esto puede proporcionar información acerca de cómo la transmisión de SARS-CoV-2 estuvo ocurriendo con medidas estrictas de distanciamiento social. Encontramos que las 5 secuencias son cercanamente relacionados a otras secuencias obtenidas en NY y CT, pero aún no es posible concluir si son el resultado de transmisión comunitaria o introducciones nuevas. Todas las 5 secuencias pertenecen al clado-NY más grande, lo cual podría indicar que el clado de NY se ha convertido en el predominante de CT; sin embargo, los 5 genomas fueron también obtenidos del área de New Haven, así que es muy probable que existan otros linajes circulando en algún otro lugar.
Conclusiones & Limitaciones
Nuestro equipo mostró en actualizaciones pasadas que los brotes en CT fueron muy probablemente iniciados por introducciones domésticas de la costa oeste y de Nueva York, variando desde Marzo hasta inicios de Abril. Nosotros demostramos que los casos del sureste de CT a finales de abril, después del pico de transmisión, fueron posiblemente más relacionados a la transmisión que ya existía en CT y NY, y no nuevas introducciones por fuentes domésticas o internacionales fuera del noreste de E.U.A.
Es importante entender que el hecho de que el clado-NY se ha convertido en el predominante en CT sureste-central no necesariamente indica que este clado es más transmisible que otros. Existe una gran cantidad de factores que podrían ser responsables de esto, como la introducción más temprana o mayor de virus de un determinado linaje, la probabilidad de la introducción de un determinado clado a una población como altos niveles de transmisión debido a factores sociales (ejemplo trabajadores esenciales, individuos sin un hogar estable, o individuos que viven en albergues, etc.), e incluso la imparcialidad del muestreo (aunque intentemos muestrear en donde que existan casos, el adquirir muestras estatales de transmisión asintomática es difícil.)
Implicaciones políticas
Nuestros resultados anteriores demuestran la importancia crucial de la cooperación intermunicipal e interestatal, y la coordinación de muestreo, rastreo de contactos, e intervenciones de distanciamiento social. Los virus se diseminan sin importar las barreras municipales o estatales, así que las estrategias de mitigación y supresión también deben de cruzar estas barreras.
Los genomas con los que contamos después del pico de transmisión son aún muy pocos para definir conclusiones, así como una fuerte imparcialidad de muestreo hacia el sureste de CT. De manera que secuenciemos más genomas, podremos definir a qué nivel es la más alta tasa de transmisión (por ejemplo dentro de las mismas ciudades, dentro de todo el estado, entre estados o introducciones internacionales). También comenzaremos a usar esta información en conjunto con los números de casos para determinar cómo las estrategias de mitigación están funcionando en un período largo, y cómo se podrían mejorar.
Metodología
Usamos 121 genomas que fueron previamente secuenciados, y secuenciamos 5 nuevos, todos usando la plataforma MinION y siguiendo el protocolo ARTIC. Para desarrollar este análisis preeliminar, también descargamos 653 genomas disponibles en GISAID, los cuales son del mundo y de E.U.A., para revelar patrones recientes de diseminación viral dentro del noreste de E.U.A. en las semanas pasadas. Los alineamientos y el análisis filogénetico de las secuencias fueron realizados usando el procedimiento nextstrain. La información geográfica de cada secuencia fue agregada por áreas del Zip Code con más de 50,000 habitantes, mayormente de CT y sus fronteras.
Disponibilidad de la información
Los directorios consensus_genomes y la información cruda en nuestro GitHub repository contienen todos nuestros genomas de SARS-CoV-2 hasta la fecha. El directorio contiene un archivo JSON que fue producido al usar el procedimiento nextstrain. Una lista de los números de acceso de los genomas de GISAID usados en este reporte se pueden encontrar en Nextstrain page.
Reconocimientos
Mary Petrone, Anne Wyllie, Chantal Vogels, Ed Courchiane, Sarah Prophet, Isabel Ott, y Chaney Kalinich realizaron las extracciones virales de RNA. Tara Alpert y Joseph Fauver prepararon las muestras para la secuenciación y ensamblaron los genomas de SARS-CoV-2. Cole Jensen y Anderson Brito realizaron el análisis filogenético. Cole Jensen, Chaney Kalinich, Mary Petrone, Anderson Brito, y Nathan Grubaugh escribieron y revisaron este reporte. Chaney Kalinich y Peter Neugebauer desarrollaron y mantienen el sitio COVIDTrackerCT. Mario Peña-Hernández lidera las traducciones al español. Nathan Grubaugh lidera la iniciativa de Vigilancia Epidemiológica y Genómica de SARS-CoV-2 de Yale. Finalmente, también agradecemos a los autores de los genomas en nuestro conjunto de datos complementario por hacer su información libremente disponible a otros investigadores: una lista completa de autores es proporcionada al final de nuestrá página dedicatoria en: nextstrain page.
Grubaugh Lab | Yale School of Public Health (YSPH) | https://grubaughlab.com/
